
The Pharmacorank Search Tool for the Retrieval of Prioritized Protein Drug Targets and Drug Repositioning Candidates According to Selected Diseases

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Overview
2.2. Assemble Protein–Disease Datasets (Overall Step 1)
2.2.1. Implement the Prioritization Algorithm Using Protein Functions (Overall Step 2, First Part)
2.2.2. Calculate the Priority Scores (Overall Step 2, Second Part)
2.3. Select Diseases Used for the Validation Studies (Overall Step 3)
2.4. Evaluate the Contributions of the Types of Functions to the Priority Score Accuracy (Overall Step 4)
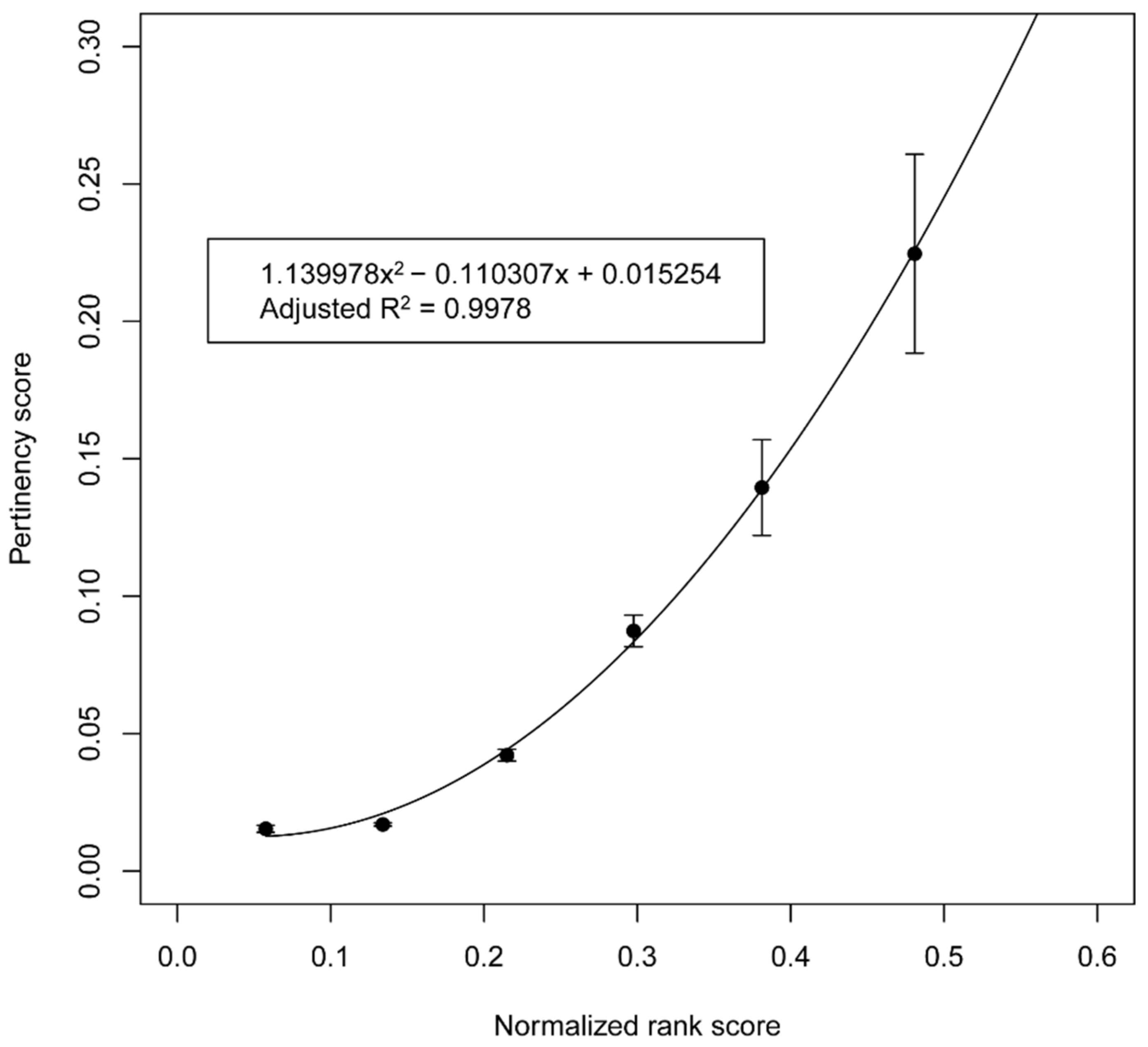
2.5. Relationship between the Priority Score and Pertinency Score (Overall Step 5 and 6)
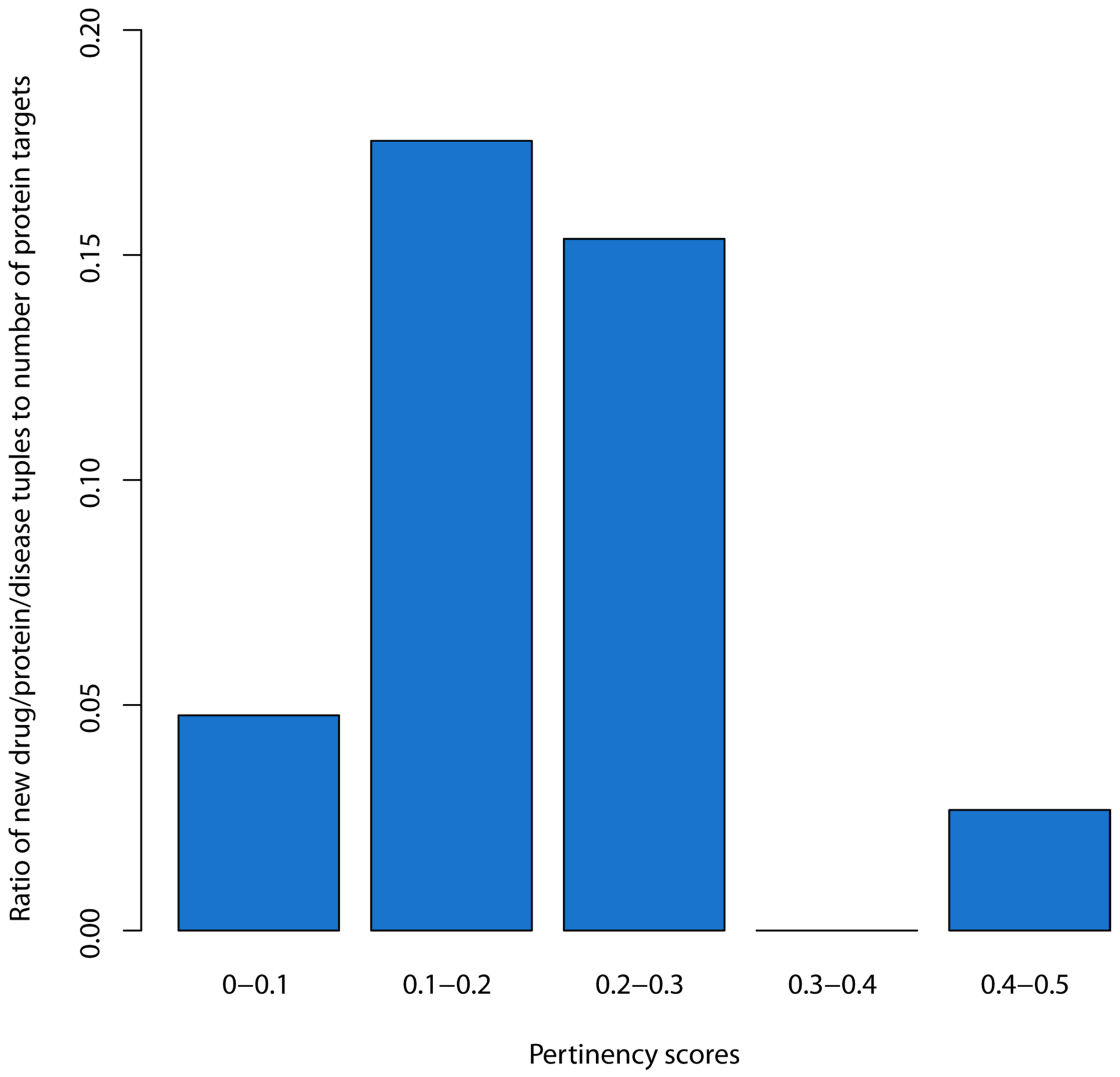
2.6. Identification of a Threshold for the Pertinency Score (Overall Step 7)
2.7. The Retrieval of Results on the Pharmacorank Site (Overall Step 8)
3. Results
3.1. Validation Studies of the Priority Score
3.2. Predictive Relationship between the Priority Score and Pertinency Score
3.3. Estimation of an Empirical Threshold for the Pertinency Score
3.4. Three Illustrative Examples of Repositioning Candidates
3.5. Examples of Repositioning Candidates for Alzheimer’s Disease
4. Discussion
4.1. Pharmacorank’s Possible Role in Enabling Drug Repositioning
4.2. Relation to Other Tools for Drug Repositioning
4.3. Significance of the Relationship between the Priority Score and Pertinency Score
4.4. Access
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McKusick, V.A. Mendelian Inheritance in Man and its online version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38 (Suppl. 1), D355–D360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32 (Suppl. 1), D115–D119. [Google Scholar] [CrossRef] [PubMed]
- Bult, C.J.; Eppig, J.T.; Kadin, J.A.; Richardson, J.E.; Blake, J.A. The Mouse Genome Database (MGD): Mouse biology and model systems. Nucleic Acids Res. 2008, 36 (Suppl. 1), D724–D728. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.J.; Zeng, K.; Kilbourne, J.; Powell, T.; Moore, R. Normalized names for clinical drugs: RxNorm at 6 years. J. Am. Med. Inform. Assoc. 2011, 18, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef] [Green Version]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2008, 36 (Suppl. 1), D919–D922. [Google Scholar] [CrossRef]
- Zhu, F.; Han, B.; Kumar, P.; Liu, X.; Ma, X.; Wei, X.; Huang, L.; Guo, Y.; Han, L.; Zheng, C. Update of TTD: Therapeutic target database. Nucleic Acids Res. 2010, 38 (Suppl. 1), D787–D791. [Google Scholar] [CrossRef] [Green Version]
- Hewett, M.; Oliver, D.E.; Rubin, D.L.; Easton, K.L.; Stuart, J.M.; Altman, R.B.; Klein, T.E. PharmGKB: The pharmacogenetics knowledge base. Nucleic Acids Res. 2002, 30, 163–165. [Google Scholar] [CrossRef]
- Degtyarenko, K.; de Matos, P.; Ennis, M.; Hastings, J.; Zbinden, M.; McNaught, A.; Alcantara, R.; Darsow, M.; Guedj, M.; Ashburner, M. ChEBI: A database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–D350. [Google Scholar] [CrossRef]
- Avram, S.; Bologa, C.G.; Holmes, J.; Bocci, G.; Wilson, T.B.; Nguyen, D.-T.; Curpan, R.; Halip, L.; Bora, A.; Yang, J.J. DrugCentral 2021 supports drug discovery and repositioning. Nucleic Acids Res. 2021, 49, D1160–D1169. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Li, J.; Xie, H.; Wang, Y. Review of drug repositioning approaches and resources. Int. J. Biol. Sci. 2018, 14, 1232. [Google Scholar] [CrossRef] [Green Version]
- Moreau, Y.; Tranchevent, L.-C. Computational tools for prioritizing candidate genes: Boosting disease gene discovery. Nat. Rev. Genet. 2012, 13, 523–536. [Google Scholar] [CrossRef]
- Yang, Y.; Adelstein, S.J.; Kassis, A.I. Target discovery from data mining approaches. Drug Discov. Today 2009, 14, 147–154. [Google Scholar] [CrossRef]
- Hurle, M.; Yang, L.; Xie, Q.; Rajpal, D.; Sanseau, P.; Agarwal, P. Computational drug repositioning: From data to therapeutics. Clin. Pharmacol. Ther. 2013, 93, 335–341. [Google Scholar] [CrossRef]
- Cheng, D.; Knox, C.; Young, N.; Stothard, P.; Damaraju, S.; Wishart, D.S. PolySearch: A web-based text mining system for extracting relationships between human diseases, genes, mutations, drugs and metabolites. Nucleic Acids Res. 2008, 36, W399–W405. [Google Scholar] [CrossRef] [Green Version]
- Julfayev, E.S.; McLaughlin, R.J.; Tao, Y.P.; McLaughlin, W.A. KB-Rank: Efficient protein structure and functional annotation identification via text query. J. Struct. Funct. Genomics 2012, 13, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Gudivada, R.C.; Aronow, B.J.; Jegga, A.G. Computational drug repositioning through heterogeneous network clustering. BMC Syst. Biol. 2013, 7 (Suppl. 5), S6. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Tripathi, L.P.; Mizuguchi, K. TargetMine, an integrated data warehouse for candidate gene prioritisation and target discovery. PLoS ONE 2011, 6, e17844. [Google Scholar] [CrossRef] [PubMed]
- Emig, D.; Ivliev, A.; Pustovalova, O.; Lancashire, L.; Bureeva, S.; Nikolsky, Y.; Bessarabova, M. Drug target prediction and repositioning using an integrated network-based approach. PLoS ONE 2013, 8, e60618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martınez, V.; Navarro, C.; Cano, C.; Blanco, A. Network-based drug-disease relation prioritization using ProphNet. In Proceedings of the International Work-Conference on Bioinformatics and Biomedical Engineering, IWBBIO 2013, Granada, Spain, 18–20 March 2013. [Google Scholar]
- Shameer, K.; Glicksberg, B.S.; Hodos, R.; Johnson, K.W.; Badgeley, M.A.; Readhead, B.; Tomlinson, M.S.; O’Connor, T.; Miotto, R.; Kidd, B.A. Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Brief. Bioinform. 2017, 19, 656–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himmelstein, D.S.; Lizee, A.; Hessler, C.; Brueggeman, L.; Chen, S.L.; Hadley, D.; Green, A.; Khankhanian, P.; Baranzini, S.E. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife 2017, 6, e26726. [Google Scholar] [CrossRef] [PubMed]
- Tranchevent, L.-C.; Capdevila, F.B.; Nitsch, D.; De Moor, B.; De Causmaecker, P.; Moreau, Y. A guide to web tools to prioritize candidate genes. Brief. Bioinform. 2011, 12, 22–32. [Google Scholar] [CrossRef] [Green Version]
- Schriml, L.M.; Mitraka, E.; Munro, J.; Tauber, B.; Schor, M.; Nickle, L.; Felix, V.; Jeng, L.; Bearer, C.; Lichenstein, R. Human Disease Ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Res. 2019, 47, D955–D962. [Google Scholar] [CrossRef] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Barrell, D.; Dimmer, E.; Huntley, R.P.; Binns, D.; O’Donovan, C.; Apweiler, R. The GOA database in 2009—An integrated Gene Ontology Annotation resource. Nucleic Acids Res. 2009, 37 (Suppl. 1), D396–D403. [Google Scholar] [CrossRef] [Green Version]
- Huntley, R.P.; Sawford, T.; Martin, M.J.; O’Donovan, C. Understanding how and why the Gene Ontology and its annotations evolve: The GO within UniProt. Gigascience 2014, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Gelman, A. Scaling regression inputs by dividing by two standard deviations. Stat. Med. 2008, 27, 2865–2873. [Google Scholar] [CrossRef]
- Wei, W.-Q.; Cronin, R.M.; Xu, H.; Lasko, T.A.; Bastarache, L.; Denny, J.C. Development and evaluation of an ensemble resource linking medications to their indications. J. Am. Med. Inform. Assoc. 2013, 20, 954–961. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; ISBN 3-900051-07-0: 2014. [Google Scholar]
- Almeida, A.M.d.; Castel-Branco, M.M.; Falcao, A.; R Core. Linear regressionR: A language and environment for calibration lines revisited: Weighting schemes for bioanalytical methods statistical computing. Vienna, Austria; 2014. J. Chromatogr. B 2002, 774, 215–222. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Groesser, L.; Herschberger, E.; Ruetten, A.; Ruivenkamp, C.; Lopriore, E.; Zutt, M.; Langmann, T.; Singer, S.; Klingseisen, L.; Schneider-Brachert, W. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat. Genet. 2012, 44, 783–787. [Google Scholar] [CrossRef]
- Green, T.E.; MacGregor, D.; Carden, S.M.; Harris, R.V.; Hewitt, C.A.; Berkovic, S.F.; Penington, A.J.; Scheffer, I.E.; Hildebrand, M.S. Identification of a recurrent mosaic KRAS variant in brain tissue from an individual with nevus sebaceous syndrome. Mol. Case Stud. 2021, 7, a006133. [Google Scholar] [CrossRef]
- Maegawa, G.H.; Tropak, M.; Buttner, J.; Stockley, T.; Kok, F.; Clarke, J.T.; Mahuran, D.J. Pyrimethamine as a potential pharmacological chaperone for late-onset forms of GM2 gangliosidosis. J. Biol. Chem. 2007, 282, 9150–9161. [Google Scholar] [CrossRef] [Green Version]
- Osher, E.; Fattal-Valevski, A.; Sagie, L.; Urshanski, N.; Sagiv, N.; Peleg, L.; Lerman-Sagie, T.; Zimran, A.; Elstein, D.; Navon, R. Effect of cyclic, low dose pyrimethamine treatment in patients with Late Onset Tay Sachs: An open label, extended pilot study. Orphanet J. Rare Dis. 2015, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Leal, A.F.; Benincore-Flórez, E.; Solano-Galarza, D.; Garzón Jaramillo, R.G.; Echeverri-Peña, O.Y.; Suarez, D.A.; Alméciga-Díaz, C.J.; Espejo-Mojica, A.J. GM2 gangliosidoses: Clinical features, pathophysiological aspects, and current therapies. Int. J. Mol. Sci. 2020, 21, 6213. [Google Scholar] [CrossRef]
- Balasubramanian, A.; Manzano, M.; Teramoto, T.; Pilankatta, R.; Padmanabhan, R. High-throughput screening for the identification of small-molecule inhibitors of the flaviviral protease. Antivir. Res. 2016, 134, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Nitsche, C. Strategies towards protease inhibitors for emerging flaviviruses. In Dengue and Zika: Control and Antiviral Treatment Strategies; Springer: Berlin/Heidelberg, Germany, 2018; pp. 175–186. [Google Scholar]
- Food, U.; Administration, D.; FDA Online Label Repository. FDA Label Search-Package Code. Available online: http://labels.fda.gov/packagecode.cfm (accessed on 3 August 2022).
- Kellar, D.; Register, T.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal insulin modulates cerebrospinal fluid markers of neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A randomized trial. Sci. Rep. 2022, 12, 1346. [Google Scholar] [CrossRef]
- Imamura, Y.; Okuzumi, A.; Yoshinaga, S.; Hiyama, A.; Furukawa, Y.; Miyasaka, T.; Hattori, N.; Nukina, N. Quantum-dot-labeled synuclein seed assay identifies drugs modulating the experimental prion-like transmission. Commun. Biol. 2022, 5, 636. [Google Scholar] [CrossRef] [PubMed]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, D.C.; Mao, X.; Dowd, K.; Tsakanikas, D.; Jiang, C.S.; Meuser, C.; Andrews, R.D.; Lukic, A.S.; Lee, J.; Hampilos, N. Riluzole, a glutamate modulator, slows cerebral glucose metabolism decline in patients with Alzheimer’s disease. Brain 2021, 144, 3742–3755. [Google Scholar] [CrossRef] [PubMed]
- Czapski, G.A.; Strosznajder, J.B. Glutamate and GABA in microglia-neuron cross-talk in Alzheimer’s disease. Int. J. Mol. Sci. 2021, 22, 11677. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, M.; Gray, J.D.; Larson, C.S.; Kazim, S.F.; Soya, H.; McEwen, B.S.; Pereira, A.C. Riluzole reduces amyloid beta pathology, improves memory, and restores gene expression changes in a transgenic mouse model of early-onset Alzheimer’s disease. Transl. Psychiatry 2018, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Rygiel, K. Can angiotensin-converting enzyme inhibitors impact cognitive decline in early stages of Alzheimer’s disease? An overview of research evidence in the elderly patient population. J. Postgrad. Med. 2016, 62, 242. [Google Scholar] [CrossRef]
- Ryan, D.K.; Karhunen, V.; Su, B.; Traylor, M.; Richardson, T.G.; Burgess, S.; Tzoulaki, I.; Gill, D. Genetic Evidence for Protective Effects of Angiotensin-Converting Enzyme Against Alzheimer Disease But Not Other Neurodegenerative Diseases in European Populations. Neurol. Genet. 2022, 8, e200014. [Google Scholar] [CrossRef]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Levinger, I.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kruzliak, P. The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef]
- Rodríguez-Puertas, R. ACE2 activators for the treatment of COVID 19 patients. J. Med. Virol. 2020, 92, 1701–1702. [Google Scholar] [CrossRef]
- Villa, C.; Rivellini, E.; Lavitrano, M.; Combi, R. Can SARS-CoV-2 infection exacerbate Alzheimer’s disease? An overview of shared risk factors and pathogenetic mechanisms. J. Pers. Med. 2022, 12, 29. [Google Scholar] [CrossRef]
- Duan, R.; Xue, X.; Zhang, Q.-Q.; Wang, S.-Y.; Gong, P.-Y.; Yan, E.; Jiang, T.; Zhang, Y.-D. ACE2 activator diminazene aceturate ameliorates Alzheimer’s disease-like neuropathology and rescues cognitive impairment in SAMP8 mice. Aging 2020, 12, 14819. [Google Scholar] [CrossRef]
- Rajapaksha, I.G.; Mak, K.Y.; Huang, P.; Burrell, L.M.; Angus, P.W.; Herath, C.B. The small molecule drug diminazene aceturate inhibits liver injury and biliary fibrosis in mice. Sci. Rep. 2018, 8, 10175. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Jones, S.J. Drug repositioning for personalized medicine. Genome Med. 2012, 4, 27. [Google Scholar] [CrossRef] [Green Version]
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef]
- Sanseau, P.; Koehler, J. Editorial: Computational methods for drug repurposing. Brief. Bioinform. 2011, 12, 301–302. [Google Scholar] [CrossRef] [Green Version]
- Paik, H.; Chung, A.-Y.; Park, H.-C.; Park, R.W.; Suk, K.; Kim, J.; Kim, H.; Lee, K.; Butte, A.J. Repurpose terbutaline sulfate for amyotrophic lateral sclerosis using electronic medical records. Sci. Rep. 2015, 5, 8580. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Aldrich, M.C.; Chen, Q.; Liu, H.; Peterson, N.B.; Dai, Q.; Levy, M.; Shah, A.; Han, X.; Ruan, X. Validating drug repurposing signals using electronic health records: A case study of metformin associated with reduced cancer mortality. J. Am. Med. Inform. Assoc. 2015, 22, 179–191. [Google Scholar] [CrossRef]
- Duran-Frigola, M.; Mateo, L.; Aloy, P. Drug repositioning beyond the low-hanging fruits. Curr. Opin. Syst. Biol. 2017, 3, 95–102. [Google Scholar] [CrossRef]
- Xie, L.; Xie, L.; Kinnings, S.L.; Bourne, P.E. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 361–379. [Google Scholar] [CrossRef]
- Taboureau, O.; Nielsen, S.K.; Audouze, K.; Weinhold, N.; Edsgärd, D.; Roque, F.S.; Kouskoumvekaki, I.; Bora, A.; Curpan, R.; Jensen, T.S. ChemProt: A disease chemical biology database. Nucleic Acids Res. 2011, 39 (Suppl. 1), D367–D372. [Google Scholar] [CrossRef]
- Ekins, S.; Williams, A.J. Finding promiscuous old drugs for new uses. Pharm. Res. 2011, 28, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Sardana, D.; Zhu, C.; Zhang, M.; Gudivada, R.C.; Yang, L.; Jegga, A.G. Drug repositioning for orphan diseases. Brief. Bioinform. 2011, 12, 346–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekins, S.; Williams, A.J.; Krasowski, M.D.; Freundlich, J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.B. Repositioned drugs: Integrating intellectual property and regulatory strategies. Drug Discov. Today Ther. Strateg. 2012, 8, 131–137. [Google Scholar] [CrossRef]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug–disease relationships for computational drug repositioning. Brief. Bioinform. 2011, 12, 303–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, E.; Athri, P. Web-based drug repurposing tools: A survey. Brief. Bioinform. 2019, 20, 299–316. [Google Scholar] [CrossRef]
- Xie, L.; Xie, L.; Bourne, P.E. Structure-based systems biology for analyzing off-target binding. Curr. Opin. Struct. Biol. 2011, 21, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Martínez, V.; Navarro, C.; Cano, C.; Fajardo, W.; Blanco, A. DrugNet: Network-based drug–disease prioritization by integrating heterogeneous data. Artif. Intell. Med. 2015, 63, 41–49. [Google Scholar] [CrossRef]
- Kissa, M.; Tsatsaronis, G.; Schroeder, M. Prediction of drug gene associations via ontological profile similarity with application to drug repositioning. Methods 2015, 74, 71–82. [Google Scholar] [CrossRef]
- Brown, A.S.; Patel, C.J. A review of validation strategies for computational drug repositioning. Brief. Bioinform. 2018, 19, 174–177. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, S.; Kaneko, M.; Narukawa, M. Approval success rates of drug candidates based on target, action, modality, application, and their combinations. Clin. Transl. Sci. 2021, 14, 1113–1122. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, S.; Zhao, H.; Wang, C.; Gao, K.; Guo, Y.; Shen, Z.; Wang, Y.; Wang, H.; Mei, X. Probucol inhibits neural cell apoptosis via inhibition of mTOR signaling pathway after spinal cord injury. Neuroscience 2016, 329, 193–200. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Function(S) Used in The Priority Score Formulation | Average AUC and Standard Deviation | p-Value of t-Test Relative to 0.5 |
|---|---|---|
| All | 0.68936 ± 0.25888 | 1.08310 × 10−49 |
| All but ChEBI and SUPERFAMILY | 0.68661 ± 0.26522 | 9.66598 × 10−47 |
| UniProt keywords only | 0.65680 ± 0.25919 | 1.22968 × 10−36 |
| GO molecular function only | 0.65522 ± 0.24997 | 3.12006 × 10−38 |
| GO biological process only | 0.65329 ± 0.26250 | 1.42324 × 10−34 |
| UniProt residue features | 0.62528 ± 0.267669 | 7.01153 × 10−24 |
| GO cellular component | 0.61153 ± 0.27507 | 1.02508 × 10−18 |
| InterPro | 0.58814 ± 0.00050 | 3.54596 × 10−14 |
| Enzyme commission (EC) number | 0.53205 ± 0.25601 | 0.00570 |
| SUPERFAMILY identifier | 0.47983 ± 0.25644 | 0.07545 |
| ChEBI | 0.42085 ± 0.25987 | 1.55528 × 10−11 |
| Medication | Target | UniProt ID | Current Use(s) | Possible Indication(s) | Pertinency Score |
|---|---|---|---|---|---|
| Sotorasib | GTPase KRas | P01116 | Non-small cell lung cancer with KRAS G12C mutation | Linear nevus sebaceous syndrome | 0.5861 |
| Pyrimethamine | Beta-hexosamididase subunit beta | P07686 | Toxoplasmosis | GM2 gangliosides | 0.4671 |
| Tolcapone | Genome polyprotein | P29990 | Parkinson’s disease | Dengue hemorraghagic fever | 0.1942 |
| Medication | Target/Pathway | Uniport ID | Current Use(s) | Pertinency Score |
|---|---|---|---|---|
| Insulin | Isoform short of insulin receptor | P06213-2 | Types 1 and 2 diabetes mellitus | 0.1666 |
| Riluzole | Alpha-synuclein | P37840 | Amyotrophic lateral sclerosis | 0.1637 |
| Diminazene aceturate (DIZE) to increase ACE2 activity | Neuroinflammation pathway of angiotensin-converting enzyme (ACE1) | P12821 | Trypanosomiasis | 0.1442 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnilopyat, S.; DePietro, P.J.; Parry, T.K.; McLaughlin, W.A. The Pharmacorank Search Tool for the Retrieval of Prioritized Protein Drug Targets and Drug Repositioning Candidates According to Selected Diseases. Biomolecules 2022, 12, 1559. https://doi.org/10.3390/biom12111559
Gnilopyat S, DePietro PJ, Parry TK, McLaughlin WA. The Pharmacorank Search Tool for the Retrieval of Prioritized Protein Drug Targets and Drug Repositioning Candidates According to Selected Diseases. Biomolecules. 2022; 12(11):1559. https://doi.org/10.3390/biom12111559
Chicago/Turabian StyleGnilopyat, Sergey, Paul J. DePietro, Thomas K. Parry, and William A. McLaughlin. 2022. "The Pharmacorank Search Tool for the Retrieval of Prioritized Protein Drug Targets and Drug Repositioning Candidates According to Selected Diseases" Biomolecules 12, no. 11: 1559. https://doi.org/10.3390/biom12111559