An Electrochemical Process Comparison of As(III) in Simulated Groundwater at Low Voltage in Mixed and Divided Electrolytic Cells

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Apparatus
2.2. The Preparation of Simulated High Arsenic Groundwater
2.3. Setup of Electrochemical Systems
3. Results and Discussion
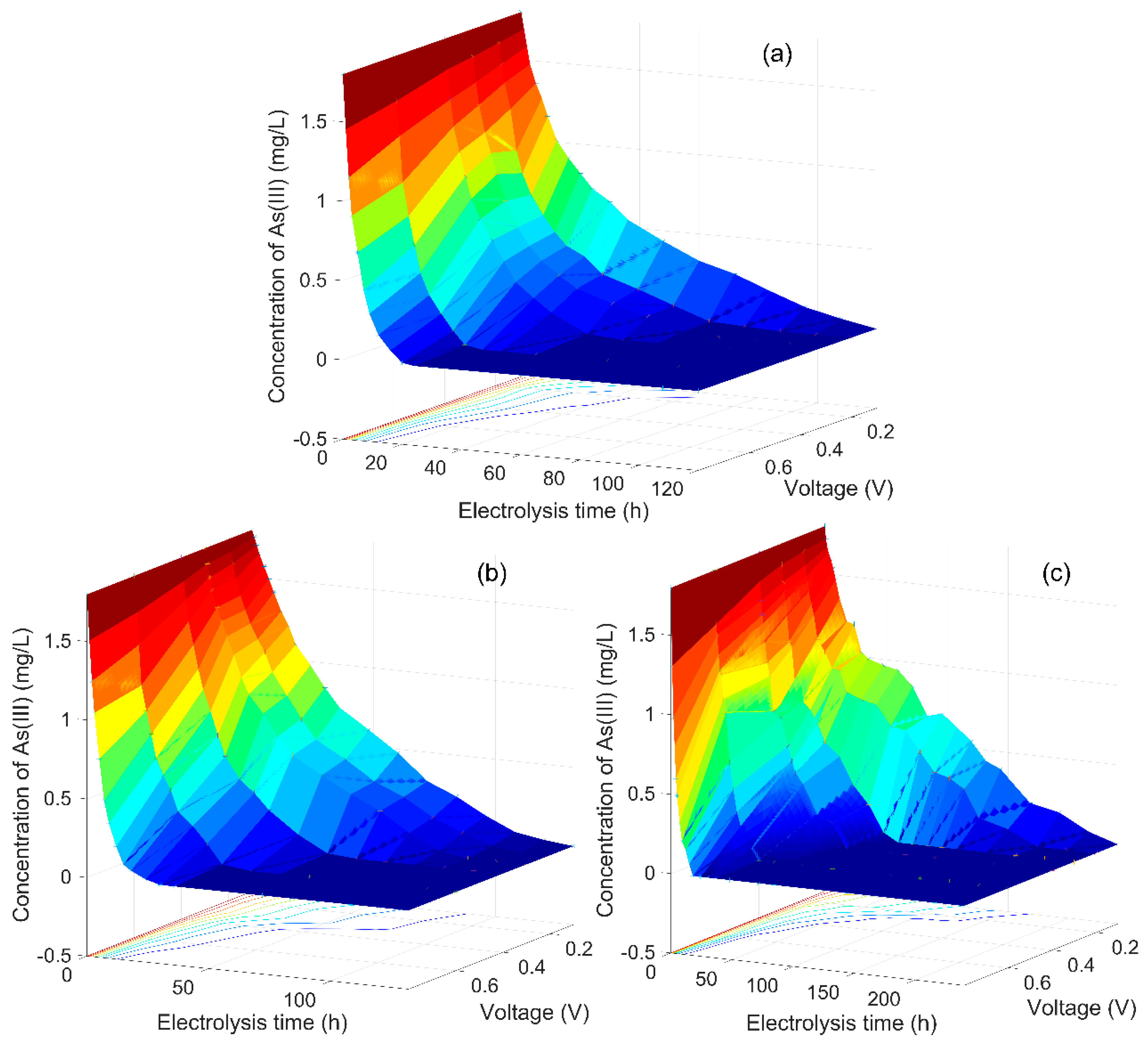
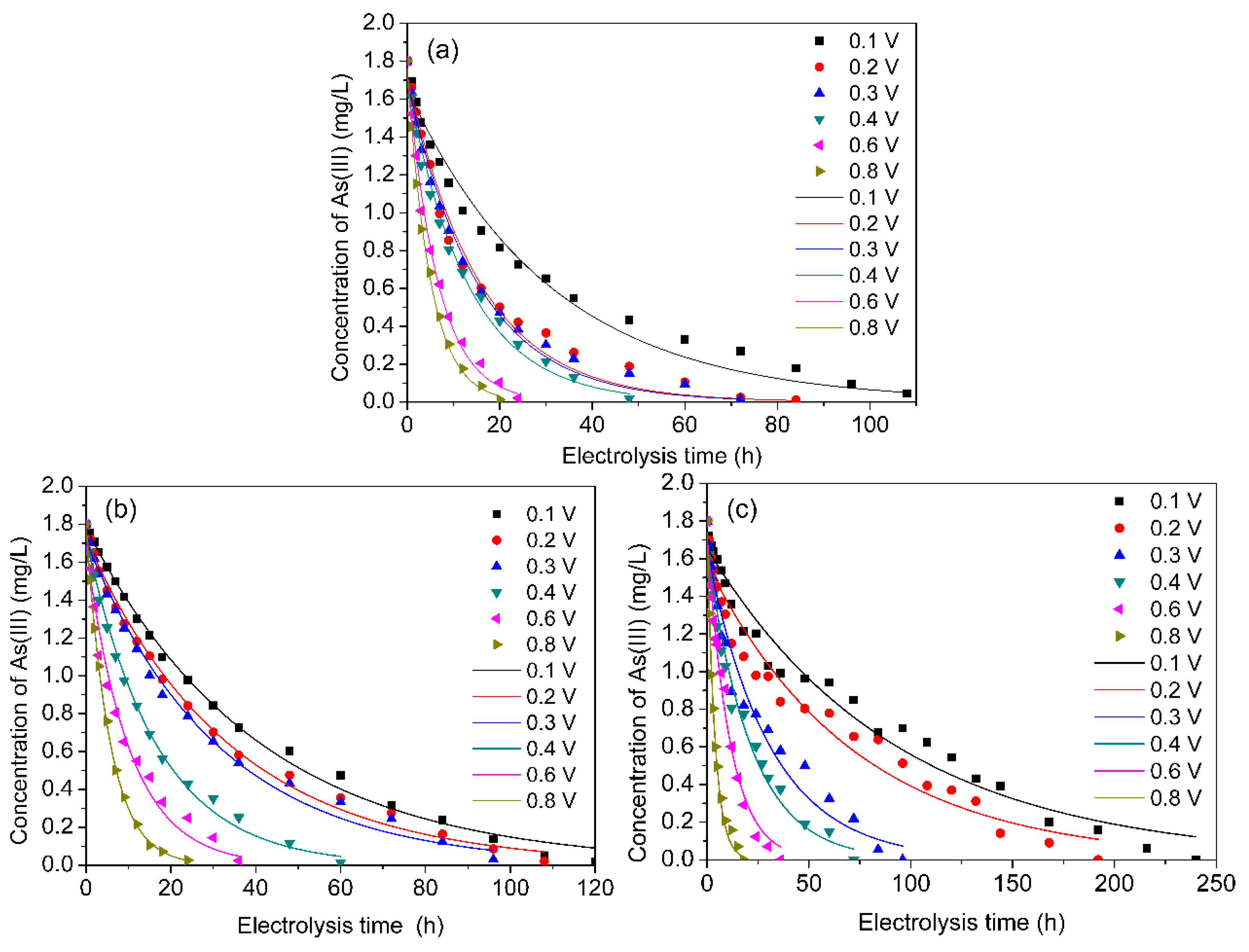
3.1. As(III) Concentration Changed with Electrolysis Time
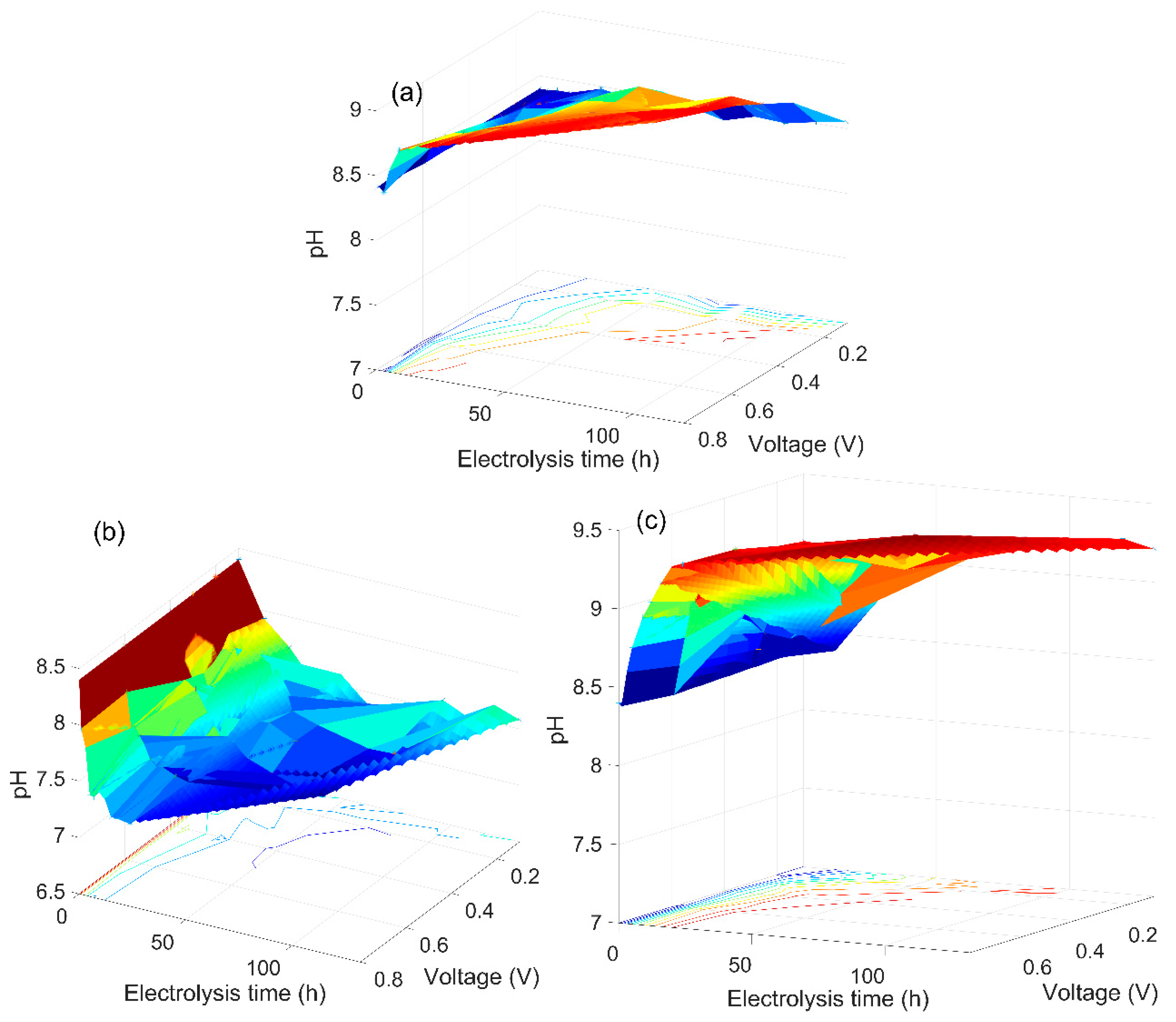
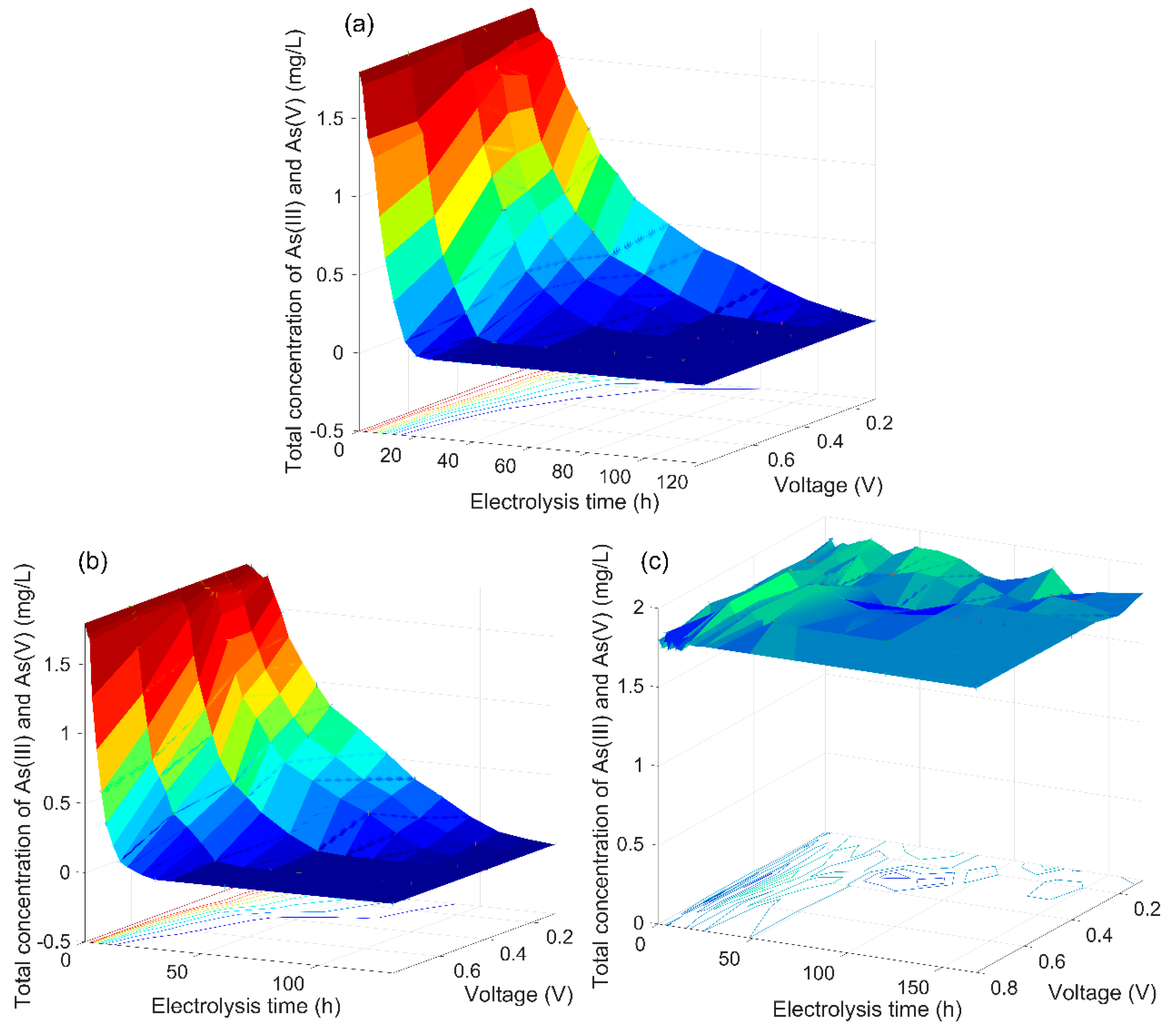
3.2. The Analysis of the As(V) Generation Process
3.3. Kinetic Study of As(III) Transformation
3.4. Analysis of Energy Consumption
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mueller, B. Arsenic in groundwater in the southern lowlands of Nepal and its mitigation options: A review. Environ. Rev. 2017, 25, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Spaziani, F.; Natori, Y.; Kinase, Y.; Kawakami, T.; Tatenuma, K. Elementary iodine-doped activated carbon as an oxidizing agent for the treatment of arsenic-enriched drinking water. Water 2019, 11, 1778. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, S.; Zhang, H.; Qiu, Y.; Zhao, J.; Zhu, Z. Decontamination of arsenic in actual water samples by calcium containing layered double hydroxides from a convenient synthesis method. Water 2018, 10, 1150. [Google Scholar] [CrossRef] [Green Version]
- Back, J.; Stadlmayr, W.; Jabornig, S.; Winkler, F.; Winkler, K.; Rupprich, M. Removal of arsenic from water with non-thermal plasma (NTP), coagulation and membrane filtration. Water 2018, 10, 1385. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Xiang, Y.; Zheng, D.; Li, Y.; Xue, S.; Wu, C.; Hartley, W.; Tan, W. Morphology-dependent enhancement of arsenite oxidation to arsenate on birnessite-type manganese oxide. Chem. Eng. J. 2017, 327, 235–243. [Google Scholar] [CrossRef]
- Zhang, P.; Tong, M.; Yuan, S.; Liao, P. Transformation and removal of arsenic in groundwater by sequential anodic oxidation and electrocoagulation. J. Contam. Hydrol. 2014, 164, 299–307. [Google Scholar] [CrossRef]
- Tong, M.; Yuan, S.; Zhang, P.; Liao, P.; Alshawabkeh, A.N.; Xie, X.; Wang, Y. Electrochemically induced oxidative precipitation of Fe(II) for As(III) oxidation and removal in synthetic groundwater. Environ. Sci. Technol. 2014, 48, 5145–5153. [Google Scholar] [CrossRef]
- Flores, O.J.; Nava, J.L.; Carreno, G.; Elorza, E.; Martinez, F. Arsenic removal from groundwater by electrocoagulation in a pre-pilot-scale continuous filter press reactor. Chem. Eng. Sci. 2013, 97, 1–6. [Google Scholar] [CrossRef]
- Maldonado-Reyes, A.; Montero-Ocampo, C.; Solorza-Feria, O. Remediation of drinking water contaminated with arsenic by the electro-removal process using different metal electrodes. J. Environ. Monit. 2017, 9, 1241. [Google Scholar] [CrossRef]
- Ratna Kumar, P.; Chaudhari, S.; Khilar, K.C.; Mahajan, S.P. Removal of arsenic from water by electrocoagulation. Chemosphere 2004, 55, 1245–1252. [Google Scholar] [CrossRef]
- Lacasa, E.; Canizares, P.; Rodrigo, M.A.; Fernandez, F.J. Electro-oxidation of As(III) with dimensionally-stable and conductive-diamond anodes. J. Hazard. Mater. 2012, 203, 22–28. [Google Scholar] [CrossRef]
- Kobya, M.; Gebologlu, U.; Ulu, F.; Oncel, S.; Demirbas, E. Removal of arsenic from drinking water by the electrocoagulation using Fe and Al electrodes. Electrochim. Acta 2011, 56, 5060–5070. [Google Scholar] [CrossRef]
- Parga, J.R.; Cocke, D.L.; Valenzuela, J.L.; Gomes, J.A.; Kesmez, M.; Irwin, G.; Moreno, H.; Weir, M. Arsenic removal via electrocoagulation from heavy metal contaminated groundwater in La Comarca Lagunera Mexico. J. Hazard. Mater. 2005, 124, 247–254. [Google Scholar] [CrossRef]
- Wan, W.; Pepping, T.J.; Banerji, T.; Chaudhari, S.; Giammar, D.E. Effects of water chemistry on arsenic removal from drinking water by electrocoagulation. Water Res. 2011, 45, 384–392. [Google Scholar] [CrossRef]
- Amrose, S.E.; Bandaru, S.R.S.; Delaire, C.; van Genuchten, C.M.; Dutta, A.; DebSarkar, A.; Orr, C.; Roy, J.; Das, A.; Gadgil, A.J. Electro-chemical arsenic remediation: Field trials in West Bengal. Environ. Int. 2009, 488–489, 539–546. [Google Scholar] [CrossRef]
- Hansen, H.K.; Nunez, P.; Jil, C. Removal of arsenic from wastewaters by airlift electrocoagulation. Part 2: Continuous reactor experiments. Sep. Sci. Technol. 2008, 43, 3663–3675. [Google Scholar] [CrossRef]
- Luo, H.; Li, C.; Wu, C.; Zheng, W.; Dong, X. Electrochemical degradation of phenol by in situ electro-generated and electro-activated hydrogen peroxide using an improved gas diffusion cathode. Electrochim. Acta 2015, 186, 486–493. [Google Scholar] [CrossRef]
- Lin, Y.; Yu, J.; Xing, Z.; Guo, X.; Yu, X.; Tang, B.; Zou, J. Enhanced generation of H2O2 and radicals on Co9S8/partly-graphitized carbon cathode for degradation of bio-refractory organic wastewater. Electrochim. Acta 2016, 213, 341–350. [Google Scholar] [CrossRef]
- Wu, Y.; Li, J.; Wang, Y.; Xie, X. Variations of uranium concentrations in a multi-aquifer system under the impact of surface water-groundwater interaction. J. Contam. Hydrol. 2018, 211, 65–76. [Google Scholar] [CrossRef]
- Lakshmanan, D.; Clifford, D.A.; Samanta, G. Ferrous and ferric ion generation during iron electrocoagulation. Environ. Sci. Technol. 2009, 43, 3853–3859. [Google Scholar] [CrossRef]
- Hug, S.J.; Leupin, O. Iron-catalyzed oxidation of arsenic(III) by oxygen and by hydrogen peroxide: pH-dependent formation of oxidants in the Fenton reaction. Environ. Sci. Technol. 2003, 37, 2734–2742. [Google Scholar] [CrossRef]
- Bataineh, H.; Oleg, P.; Andreja, B. pH-induced mechanistic changeover from hydroxyl radicals to iron(iv) in the Fenton reaction. Chem. Sci. 2012, 72, 1869–1878. [Google Scholar] [CrossRef]
- Pang, S.; Jiang, J.; Ma, J. Oxidation of sulfoxides and arsenic(III) in corrosion of nanoscale zero valent iron by oxygen: Evidence against ferryl ions (Fe(IV)) as active intermediates in Fenton reaction. Environ. Sci. Technol. 2011, 45, 307–312. [Google Scholar] [CrossRef]
- Vik, E.A.; Carlson, D.A.; Eikum, A.S.; Gjessing, E.T. Electrocoagulation of potable water. Water Res. 1984, 18, 1355–1360. [Google Scholar] [CrossRef]
- Arienzo, M.; Adamo, P.; Chiarenzelli, J.; Bianco, M.R.; De Martino, A. Retention of arsenic on hydrous ferric oxides generated by electrochemical peroxidation. Chemosphere 2002, 48, 1009–1018. [Google Scholar] [CrossRef]
- Edwards, M. Chemistry of arsenic removal during coagulation and Fe-Mn oxidation. J Am Water Works Assoc. 1994, 86, 64–78. [Google Scholar] [CrossRef]
- Li, N.; Wang, S.; An, J.; Feng, Y. Acid pretreatment of three-dimensional graphite cathodes enhances the hydrogen peroxide synthesis in bioelectrochemical systems. Sci. Total Environ. 2018, 630, 308–313. [Google Scholar] [CrossRef]
- Leng, W.H.; Zhu, W.C.; Ni, J.; Zhang, Z.; Zhang, J.Q.; Cao, C.N. Photoelectrocatalytic destruction of organics using TiO2 as photoanode with simultaneous production of H2O2 at the cathode. Appl. Catal. A Gen. 2006, 300, 24–35. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.L. The cooperative electrochemical oxidation of chlorophenols in anode–cathode compartments. J. Hazard. Mater. 2008, 154, 44–50. [Google Scholar] [CrossRef]
- Bensadok, K.; Benammar, S.; Lapicque, F.; Nezzal, G. Electrocoagulation of cutting oil emulsions using aluminium plate electrodes. J. Hazard. Mater. 2008, 152, 423–430. [Google Scholar] [CrossRef]
- Min, B.K.; Cheng, S.A.; Logan, B.E. Electricity generation using membrane and salt bridge microbial fuel cells. Water Res. 2005, 39, 1675–1686. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; van Genuchten, C.M.; Addy, S.E.A.; Yao, J.; Gao, N.; Gadgil, A.J. Modeling As(III) oxidation and removal with iron electrocoagulation in groundwater. Environ. Sci. Technol. 2012, 46, 12038–12045. [Google Scholar] [CrossRef] [PubMed]
- Secula, M.S.; Cagnon, B.; de Oliveira, T.F.; Chedeville, O.; Fauduet, H. Removal of acid dye from aqueous solutions by electrocoagulation/GAC adsorption coupling: Kinetics and electrical operating costs. J. Taiwan Inst. Chem. Eng. 2012, 43, 767–775. [Google Scholar] [CrossRef]
- Maher, E.K.; O’Malley, K.N.; Heffron, J.; Huo, J.; Mayer, B.K.; Wang, Y.; McNamara, P.J. Analysis of operational parameters, reactor kinetics, and floc characterization for the removal of estrogens via electrocoagulation. Chemosphere 2019, 220, 1141–1149. [Google Scholar] [CrossRef]
- Zhang, X.; Lei, L.; Xia, B.; Zhang, Y.; Fu, J. Oxidization of carbon nanotubes through hydroxyl radical induced by pulsed O2 plasma and its application for O2 reduction in electro-Fenton. Electrochim. Acta 2009, 54, 2810–2817. [Google Scholar] [CrossRef]
- Gong, Y.; Li, J.; Zhang, Y.; Zhang, M.; Tian, X.; Wang, A. Partial degradation of levofloxacin for biodegradability improvement by electro-Fenton process using an activated carbon fiber felt cathode. J. Hazard. Mater. 2016, 304, 320–328. [Google Scholar] [CrossRef]
- Shafaei, A.; Rezaie, M.; Nikazar, M. Evaluation of Mn2+ and Co2+ removal by electrocoagulation: A case study. Chem. Eng. Process. Process Intensif. 2011, 50, 1115–1121. [Google Scholar] [CrossRef]
- Chen, X.; Ren, P.; Li, T.; Trembly, J.P.; Liu, X. Zinc removal from model wastewater by electrocoagulation: Processing, kinetics and mechanism. Chem. Eng. J. 2018, 349, 358–367. [Google Scholar] [CrossRef]
- Yu, F.; Chen, Y.; Pan, Y.; Yang, Y.; Ma, H. A cost-effective production of hydrogen peroxide via improved mass transfer of oxygen for electro-Fenton process using the vertical flow reactor. Sep. Purif. Technol. 2020, 241, 116695. [Google Scholar] [CrossRef]
- Ghalebizade, M.; Ayati, B. Acid Orange 7 treatment and fate by electro-peroxone process using novel electrode arrangement. Chemosphere 2019, 235, 1007–1014. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrochemical Parameters | Min (mg/L) | Median (mg/L) | Mean (mg/L) | Max (mg/L) |
|---|---|---|---|---|
| Cl− | 4 | 119 | 432 | 7597 |
| NO3− | <0.05 | 17.3 | 75 | 1900 |
| SO42− | <0.05 | 129 | 396 | 6951 |
| HCO3− | 46 | 413 | 487 | 1510 |
| Ca2+ | 4.6 | 42.5 | 60.8 | 465 |
| K+ | 0.6 | 3.3 | 14.1 | 370 |
| Mg2+ | 6.3 | 43.8 | 101 | 1530 |
| Na+ | 7.8 | 169 | 335 | 3848 |
| TDS | 172 | 779 | 1658 | 20576 |
| MgSO4 | NaHCO3 | NaNO3 | KCl | CaCl2 | FeCl2 | NaCl | |
|---|---|---|---|---|---|---|---|
| Simulated groundwater (mmol/L) | 4.14 | 7.98 | 1.21 | 0.36 | 1.52 | 0.054 | 8.65 |
| Voltage (V) | The Mixed Electrolysis System | The Anodic Cell of the Separated System | The Cathodic Cell of the Separated System | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C0 (mg/L) | Kapp | R2 | C0 (mg/L) | Kapp | R2 | C0 (mg/L) | Kapp | R2 | |
| 0.1 | 1.615 | 0.029 | 0.980 | 1.892 | 0.028 | 0.987 | 1.604 | 0.010 | 0.924 |
| 0.2 | 1.644 | 0.054 | 0.979 | 1.854 | 0.033 | 0.983 | 1.516 | 0.013 | 0.919 |
| 0.3 | 1.614 | 0.057 | 0.985 | 1.792 | 0.034 | 0.988 | 1.617 | 0.031 | 0.958 |
| 0.4 | 1.835 | 0.083 | 0.976 | 1.906 | 0.068 | 0.976 | 1.658 | 0.046 | 0.977 |
| 0.6 | 1.892 | 0.163 | 0.986 | 1.738 | 0.098 | 0.978 | 1.715 | 0.088 | 0.983 |
| 0.8 | 1.981 | 0.223 | 0.976 | 1.786 | 0.179 | 1.000 | 1.735 | 0.250 | 0.992 |
| Voltage (V) | The Mixed System | The Separated System | ||||
|---|---|---|---|---|---|---|
| Charge Quantity (C) | Work (J) | Unit Energy Consumption of As(III) Conversion (J/mg) | Charge Quantity (C) | Work (J) | Unit Energy Consumption of As(III) Conversion (J/mg) | |
| 0.1 | 66.60 | 6.66 | 7.40 | 88.56 | 8.86 | 9.84 |
| 0.2 | 89.28 | 17.86 | 19.84 | 109.8 | 21.96 | 24.40 |
| 0.3 | 93.02 | 27.91 | 31.01 | 76.70 | 23.01 | 25.56 |
| 0.4 | 119.16 | 47.66 | 52.96 | 77.20 | 30.88 | 34.31 |
| 0.6 | 136.49 | 81.90 | 91.00 | 99.61 | 59.77 | 66.41 |
| 0.8 | 141.10 | 112.88 | 125.42 | 119.23 | 95.39 | 105.99 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, Y.; Cui, Y.; Lei, L.; Gao, Y.; Zhou, Z.; Li, Y.; Shi, X. An Electrochemical Process Comparison of As(III) in Simulated Groundwater at Low Voltage in Mixed and Divided Electrolytic Cells. Water 2020, 12, 1126. https://doi.org/10.3390/w12041126
Qin Y, Cui Y, Lei L, Gao Y, Zhou Z, Li Y, Shi X. An Electrochemical Process Comparison of As(III) in Simulated Groundwater at Low Voltage in Mixed and Divided Electrolytic Cells. Water. 2020; 12(4):1126. https://doi.org/10.3390/w12041126
Chicago/Turabian StyleQin, Yanyan, Yanping Cui, Lidan Lei, Ya Gao, Zhengwei Zhou, Yilian Li, and Xiaoyan Shi. 2020. "An Electrochemical Process Comparison of As(III) in Simulated Groundwater at Low Voltage in Mixed and Divided Electrolytic Cells" Water 12, no. 4: 1126. https://doi.org/10.3390/w12041126