3.1. Sampling and Analysis
On the existing monitoring network, 22 wells were sampled to analyze chlorinated solvents and among those wells, 13 were also sampled for a more detailed hydrochemical characterization of groundwater. Wells are screened in the upper part of the chalk except for the well ‘S’ equipped with a double casing allowing two sampling levels (25 m and 40 m below surface).
The sampling procedure was carried out by pumping at least 3 times the volume of water in the sampled well and monitoring of temperature, pH, electrical conductivity, redox potential, and dissolved oxygen. All these parameters were monitored using a multi probe handheld meter (WTW multi 350i, Weilheim, Germany) except dissolved oxygen that was measured by a luminescent DO probe (Hach Lange, Düsseldorf, Germany).
Samples for CAHs analyses were collected in 40 mL glass vials filled and acidified with sulphuric acid (H2SO4) at pH ≈ 2. 180 mL polypropylene bottles were used for the analysis in the lab of a standard package of major elements (Ca2+, K+, Mg2+, Na+, Cl−, SO42−, HCO3−) analyses and several minor chemical compounds (NH4+, Li+, Sr2+, NO3−, PO43−, Br−, F− and SiO2). 100 mL bottles were filled using 0.45 μm filter and acidified (37% HCl) to analyze dissolved iron and manganese. Before being analyzed, all samples were kept refrigerated at 4 °C and protected from light.
At the laboratory, the chemical analyses were carried out using the following methods:
Ion chromatography for K+, Mg2+, Na+, NH4+, Li+, Sr2+, PO43−, Br− Cl−, F−, NO3− and SO42−;
Titrimetric method for Ca2+;
Flame atomic absorption for Fe3+, Mn2+ and SiO2;
The Carbonate speciation between CO
2, HCO
3−, CO
32− is obtained from pH and total alkanity; according to Rodier’s formula [
30];
Gas chromatography coupled with mass spectrometry (GC/MS) for the CAHs.
For the organic CAHs compounds, results show that three CAHs are dominant: 1,1,1-TCA, 1,1-DCE and TCE, with maximum concentrations of 1100 μg/L, 820 μg/L and 550 μg/L respectively. Concentrations have decreased of 85.13%, 72.5% and 80.47% respectively compared March 2013, before the beginning of remediation operations of the CAHs source. Other aliphatic hydrocarbons such as 1,1,2-TCA, tetrachloromethane, 1,2-dichloroethane and cis-1,2-DCE are detected with lower concentrations. Results showed also a decrease of concentrations of all CAHs in groundwater compared to the concentrations observed in March 2013.
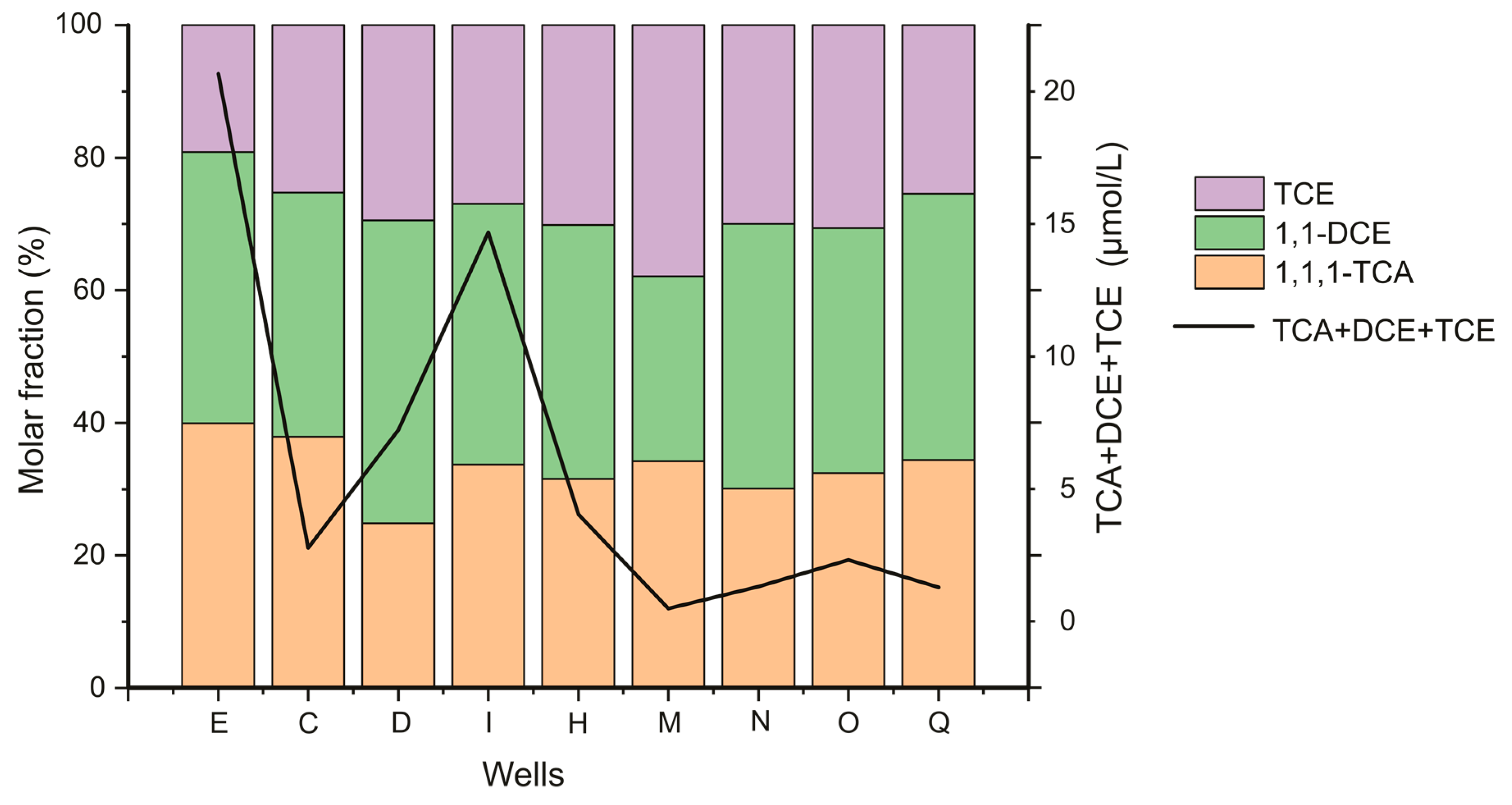
Along the plume centerline, the sum of molar concentrations of the three dominant CAHs are presented in a same graph with the molar percentage of each (
Figure 2) Despite the general decrease in CAHs concentrations, their spatial distribution is quite similar to that from Palau et al. (2016) obtained in March 2013 on this site. Along the centerline, concentrations (of the sum of 1,1,1-TCA + 1,1-DCE + TCE) show a decreasing trend from 20.66 µmol/L at well (E) to 1.28 µmol/L at well (Q). For almost all wells along the plume centerline, 1,1-DCE molar fraction is closer to the one of 1,1,1-TCA, with a slightly lower fraction for TCE.
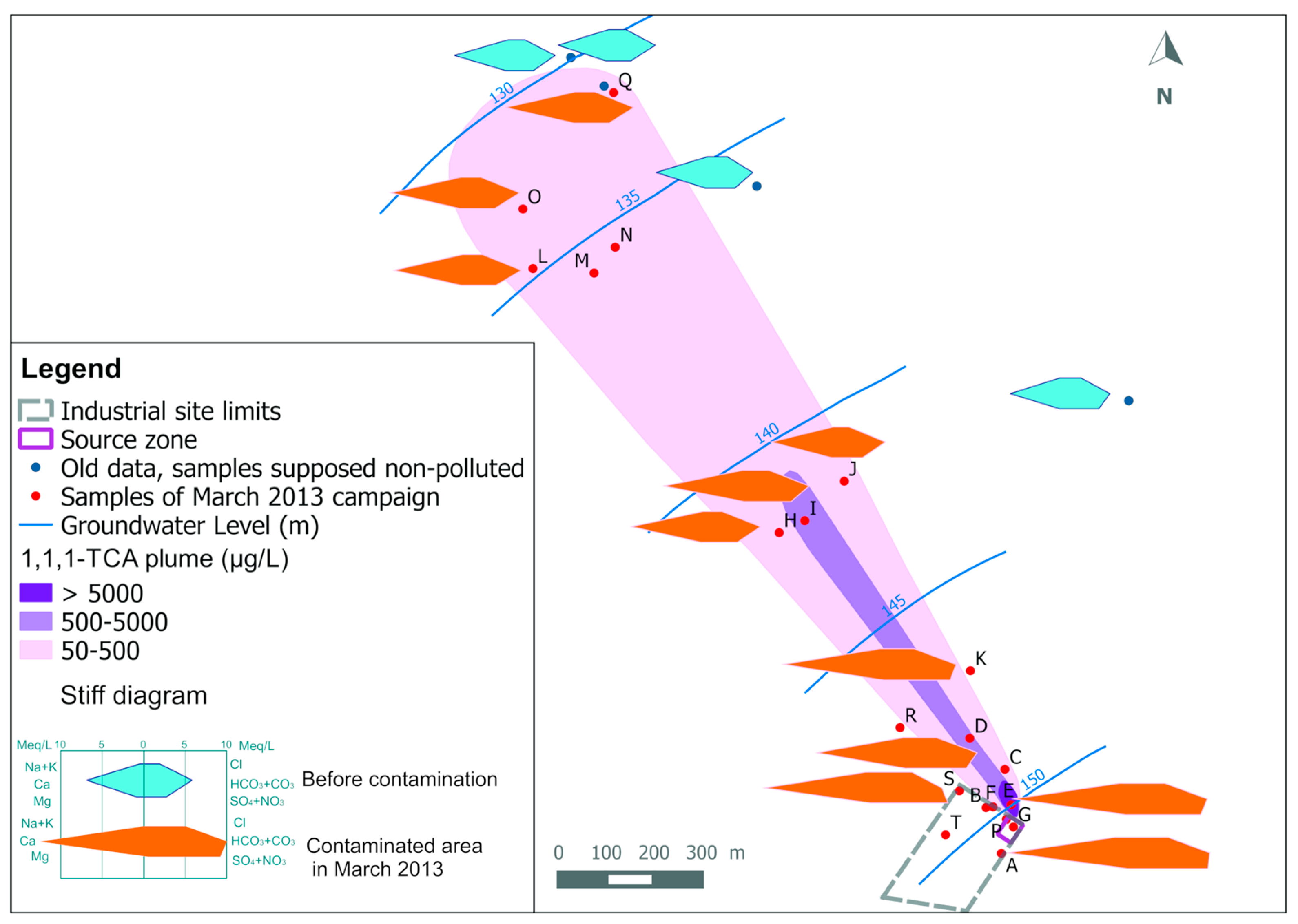
Results of chemical analyses of the inorganic elements in March 2017 are given in
Table 2. Wells are listed according to the increasing distance from the source of the CAHs. Well labels are shown in
Figure 1.The measured average temperature is 11.90 °C. The presence of dissolved oxygen with concentrations between 1.04 mg/L and 4.71 mg/L confirms the dominance of aerobic conditions in the aquifer. At most of the sampled points, the in situ measured pH is lower than 7. These values are mainly explained by HY/DH degradation reactions of 1,1,1-TCA that slightly increase water acidity due to the release of hydrochloric and acetic acids. The electrical conductivity is relatively high, ranging from 787 to 1847 µs/cm. Concentrations in major ions such as Ca
2+, HCO
3−, SO
42− and Cl
− decrease downgradient along the groundwater flow direction. Na
2+ and Mg
2+ concentrations also show a decrease, but their concentrations are relatively low. Ca
2+ concentrations decrease from 310.09 mg/L to 168.39 mg/L, downgradient from the source. Cl
− concentrations also decrease from 160.97 mg/L to 48.30 mg/L in the plume. Near the source area, the SO
42− concentration exceeds the drinking water limit of 250 mg/L set by the European Directive [
22]. It decreases from 343.63 mg/L at the well (G) to 93.06 mg/L at the well (Q) located farthest from the source area. HCO
3− concentrations are decreasing from 528.95 to 298.28 mg/L with a spatial distribution quite similar to Ca
2+ concentrations. NO
3− concentrations are higher than 50 mg/L in all sampled wells with a different spatial distribution than for the other elements. NO
3− has a distinct origin, related to intense agricultural activities with fertilizers that diffusely impact this aquifer [
31]. Compared to data before remediation (March 2013), a general slight decrease in concentrations is observed for Ca
2+, HCO
3−, SO
42−, Cl
−, Na
+ and Mg
2+.
Changes of physicochemical properties and concentrations can be investigated using different groundwater characteristics and tools (i.e., diagrams, graphs) (e.g., [
32,
33]). From our results, calcite SI were calculated with PHREEQC, using the PHREEQC database. They range between −0.58 and 0.52, indicating that groundwater can be considered in equilibrium with calcite.
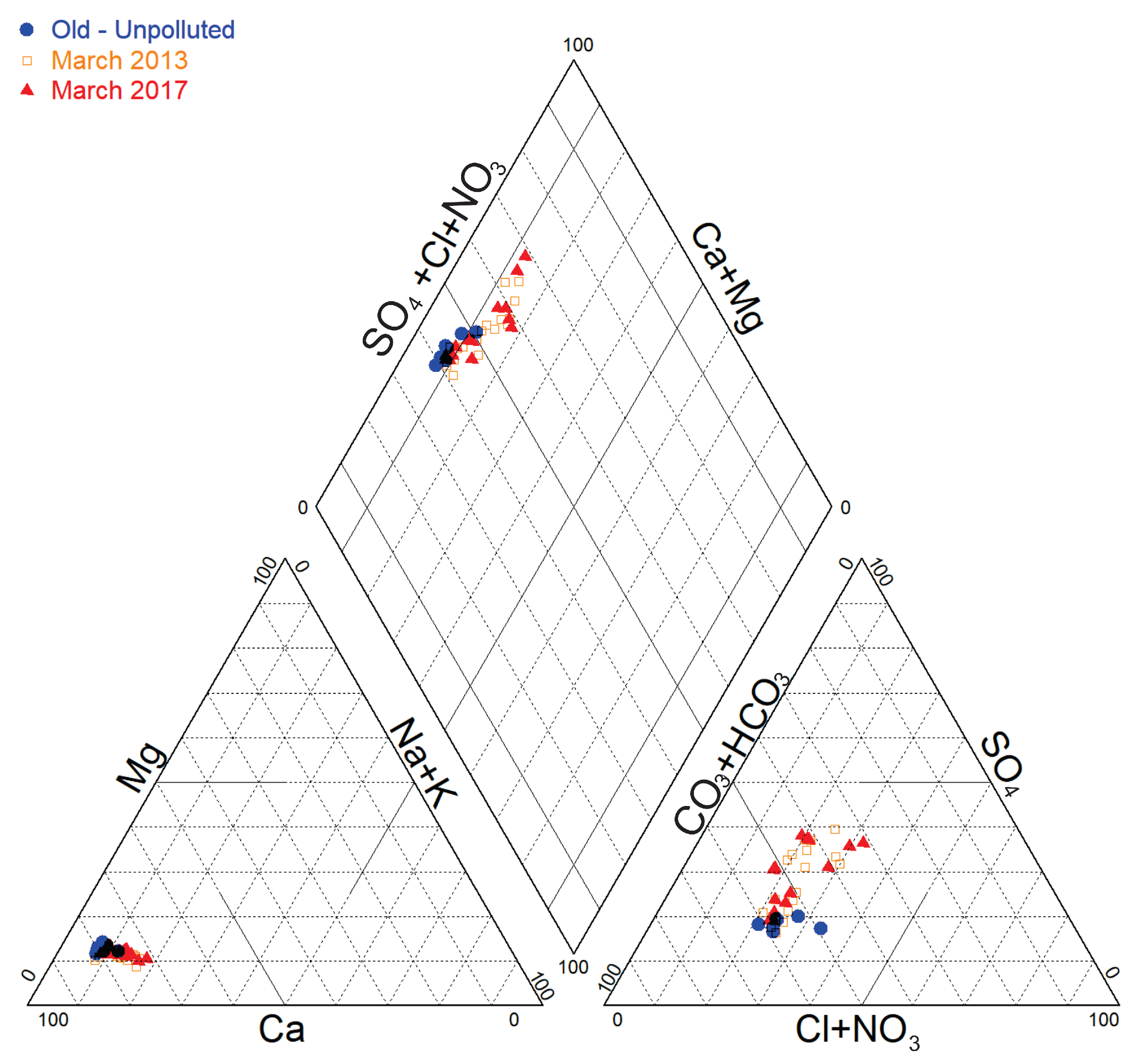
In Piper diagram (
Figure 3), a comparison of the relative compositions of groundwater was performed based on data: before pollution detection (October 1993), before source remediation (March 2013) and after source remediation (March 2017).
Historical groundwater data (reflecting unpolluted groundwater status) in the vicinity of the site show typical Ca-HCO3 hydro-chemical facies. The situation in March 2013, at the industrial site and in the direct surroundings the showed that groundwater composition was evolving progressively towards a Ca-SO4 facies, with a clear SO42− enrichment. At the same period, the groundwater composition was tending progressively back to its initial (unpolluted) composition further downgradient. In March 2017, after the CAHs source remediation, the relative chemical composition in Piper diagram seems almost similar to that of March 2013.
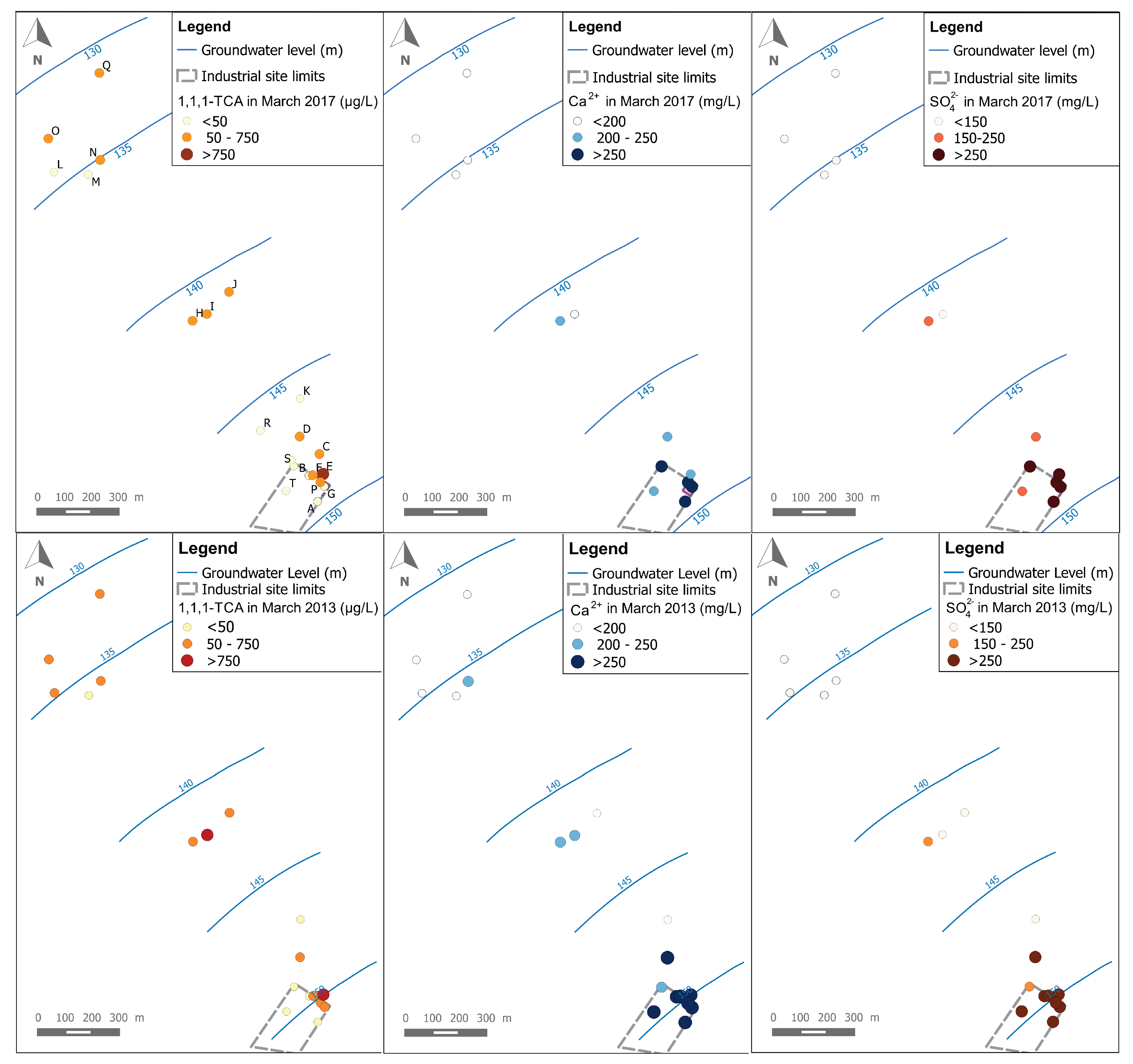
Figure 4 shows a comparison between 1,1,1-TCA, Ca
2+ and SO
42− spatial distributions for both campaigns of March 2013 and March 2017. As a first observation, the spatial distribution of 1,1,1-TCA (like other CAHs) is quite different than for Ca
2+ and SO
42−. It can be seen mainly at the industrial site where the highest concentrations of 1,1,1-TCA are detected in the north-eastern part of the industrial site (around well E), while the highest concentrations of Ca
2+ and SO
42− are observed in wells: A, B, F, G, P and S of the industrial site (i.e., where a backfill layer exists). This could be a sign of a difference between the source zone of the CAHs plume, and the actual source area of the Ca
2+ and SO
42− plumes. This observation is confirmed by data from both campaigns (i.e., 2013 and 2017).
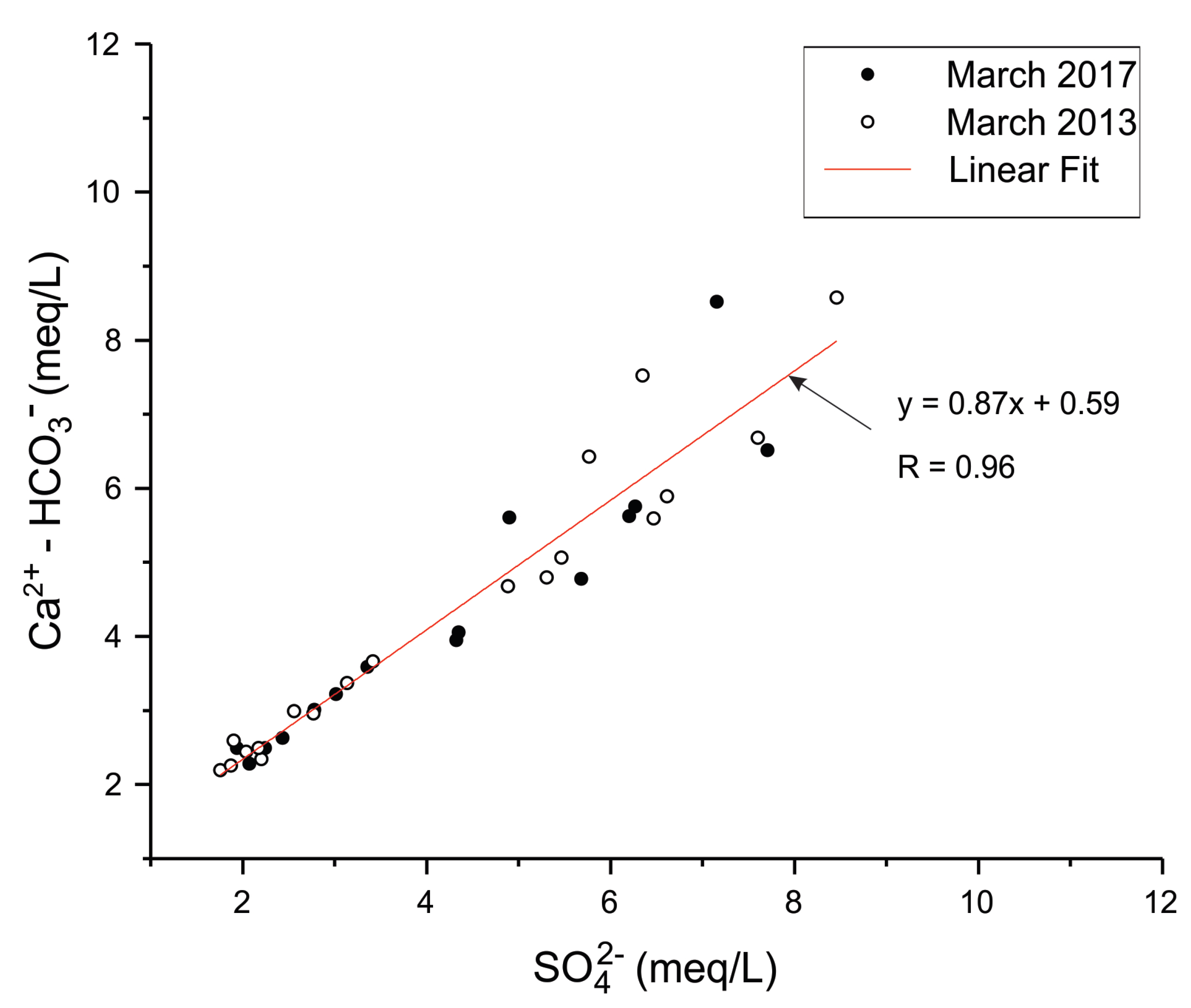
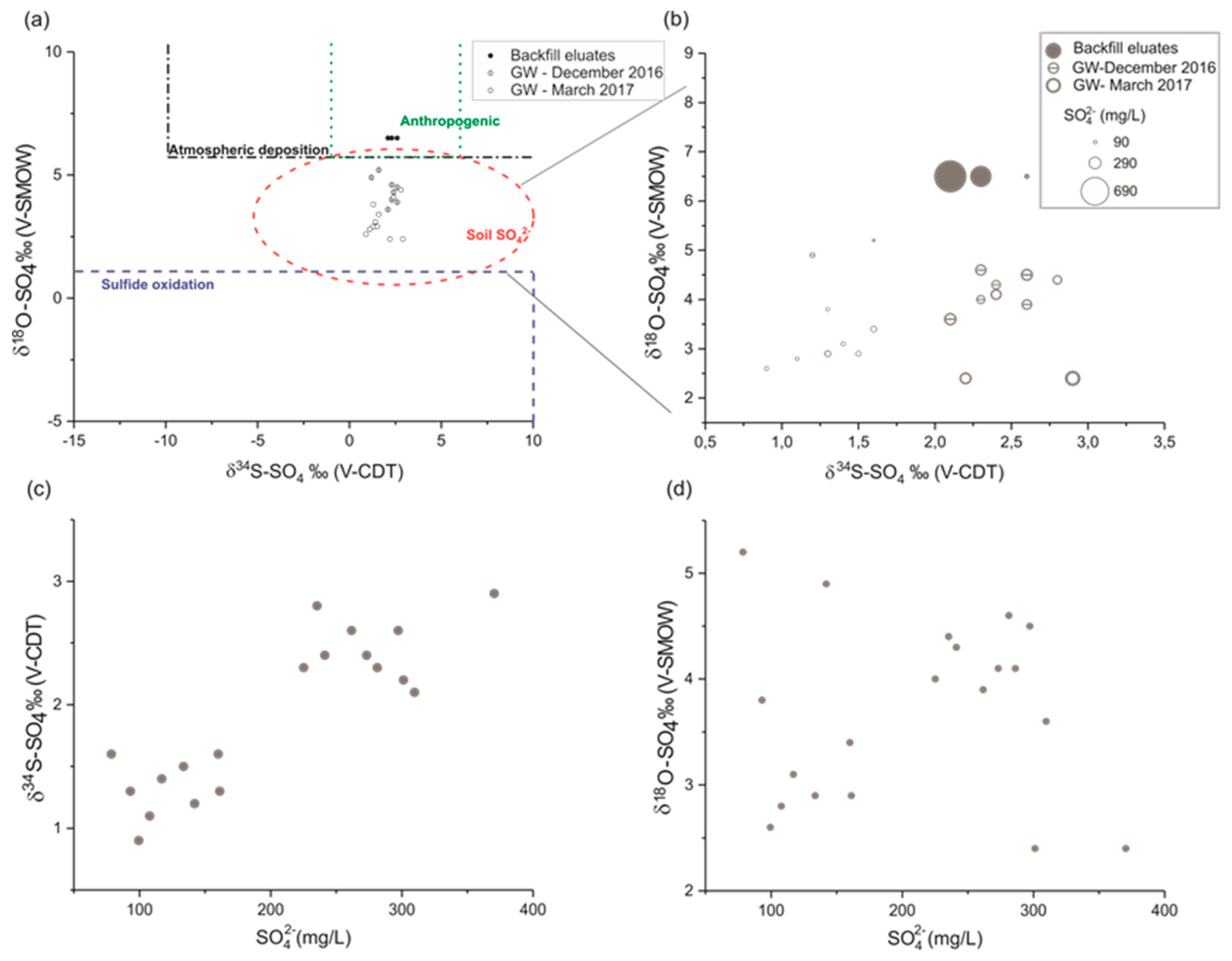
In order to confirm the calcium sulphate input in the groundwater, and to distinguish it from calcite dissolution in the chalk aquifer, both data of March 2013 and March 2017 were used to analyze the evolution of non-carbonated calcium (Ca
2+-HCO
3−) concentrations as a function of SO
42− concentrations (
Figure 5). A similar approach was used by Kimblin [
34] to investigate gypsum dissolution gypsum in the aquifer of Triassic Sherwood Sandstone Group (UK). The alignment of data points along a line close to the 1:1 line indicates a direct supply of calcium sulphate to the aquifer.
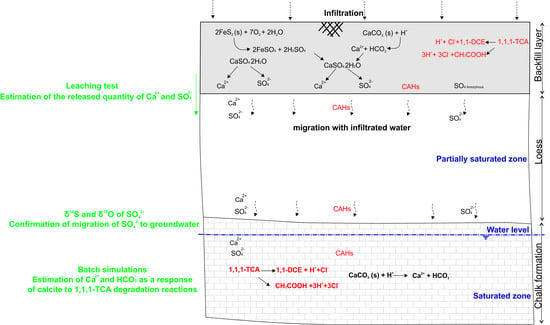
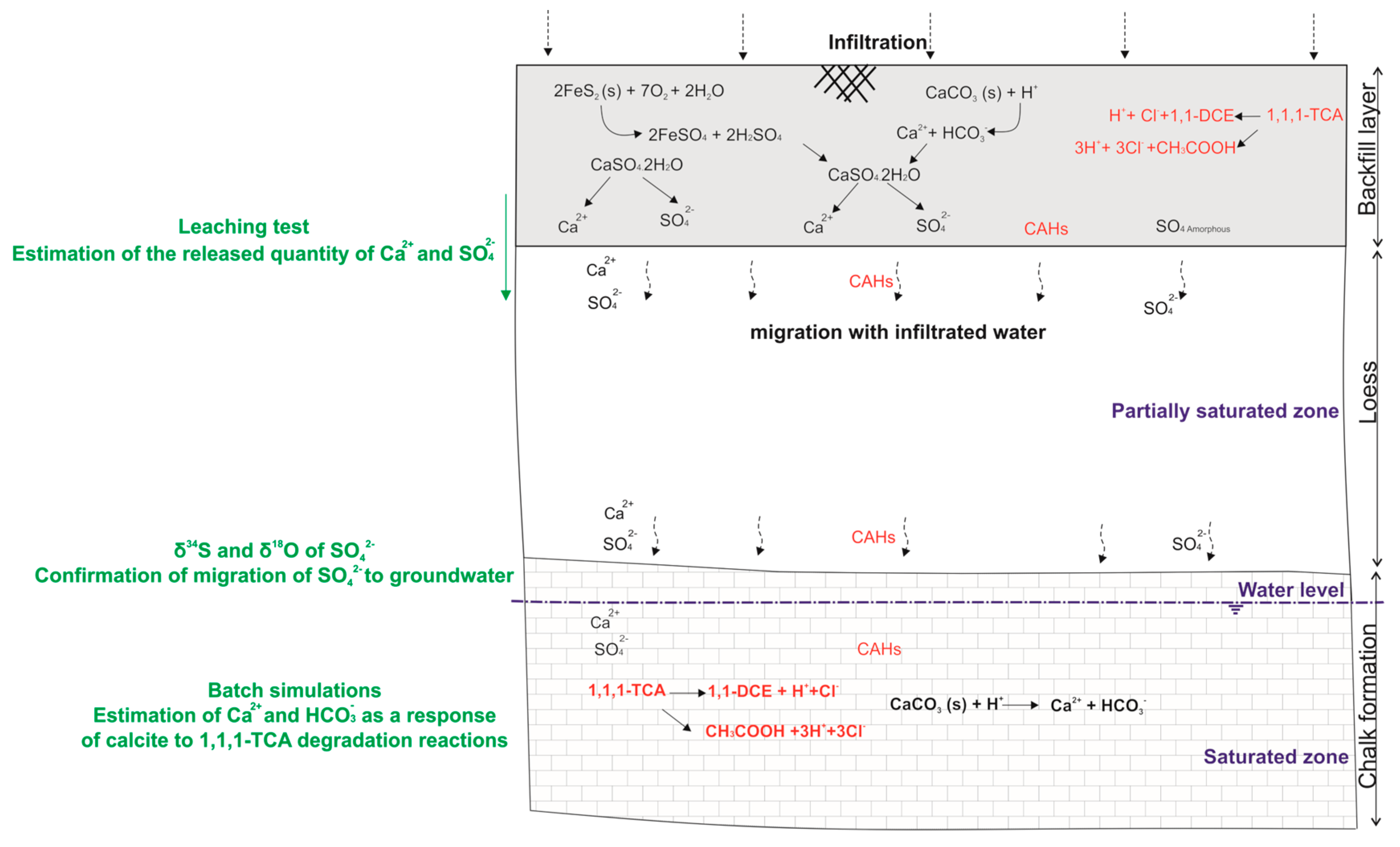
The presence of a backfill layer at the industrial site, above the observation wells that show the highest concentrations of sulphate and calcium, indicates that the backfill is the most probable source of the external input of calcium sulphate in groundwater.
3.2. Simulation of Calcite Dissolution under Degradation Reactions
Geochemical batch simulations were performed to quantify the increase of calcite dissolution in the presence of 1,1,1-TCA degradation reactions:
these two reactions show different yields due to the kinetics of HY, which is about 2.7 times faster than that of DH [
35]. According to Palau et al. [
36], the DH yield is about 27%. By using it with the molar balance, 1 mol of 1,1,1-TCA produces about 2.46 mol of HCl.
On the other hand, the degradation of 1,1,1-TCA by HY/DH are influenced by temperature [
37]. Field temperatures enable the estimation of 1,1,1-TCA degradation rate according to Arrhenius equation:
where
k is the first order degradation constant (s
−1),
A is the frequency factor (s
−1),
R is the perfect gas constant (8.314 × 10
−3 kJ mol
−1K
−1),
Ea is the activation energy (kJ mol
−1) and T the temperature (K). From Gauthier and Murphy [
38], the constants
Ea and
A are assumed to be 122.8 kJ mol
−1 and 8.7 × 10
13 s
−1 respectively. These values were obtained from several previous studies.
The quantity of HCl released in groundwater can be estimated for a given degradation time, using observed field concentrations, the reaction yields of Palau et al. [
36], and the degradation rate obtained by the Arrhenius equation.
In order to quantify Ca
2+ and HCO
3− required to reach the equilibrium with calcite, the chemical composition of an assumed unpolluted sample is used by adding the estimated HCl quantity. Then, the solution is assembled with the calcite assuming equilibrium conditions in PHREEQC [
29].
The half-life time was calculated from the Arrhenius equation (Equation (3)) with an average temperature of 11.92 °C obtained from observations in March 2017 (n = 27). The half-life time is 11.57 years, this means that for 1 year, 4.3% of the amount of 1,1,1-TCA degrades via HY/DH. The estimated groundwater pH changes with degradation of 1,1,1-TCA by HY/DH is summarized in
Table 3.
Simulation results (
Table 4) show that, for a degradation over a period of 1 year, and in the presence of only 200 µg/L of 1,1,1-TCA, the pH decreases by 0.46 units from the reference composition. Consequently, a supplement of 17.65 mg/L calcium and 53.64 mg/L bicarbonate must be released to maintain the solution in equilibrium with calcite. A concentration of 500 µg/L can decrease the pH by 0.75 unit and the equilibrium with calcite is obtained with a release of 48.14 mg/L of calcium and 146.50 mg/L of bicarbonate. To reach equilibrium with calcite for the maximum concentration 1100 µg/L of 1,1,1-TCA (observed in March 2017), the pH decreases by 0.98 unit and the dissolution of calcite increases groundwater concentrations of calcium by 95.28 mg/L and bicarbonates by 289.93 mg/L.
Using 1,1,1-TCA concentrations with observed field temperatures, these simplified simulations provide an estimation of Ca2+ and HCO3− releases in the presence of HY/DH of 1,1,1-TCA in the chalk aquifer.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}